Раздел 1
БИОЛОГИЧЕСКИЕ ОСНОВЫ ЖИЗНЕДЕЯТЕЛЬНОСТИ ЧЕЛОВЕКА
1.3. Онтогенетический уровень
организации жизни
1.3.2. Основы генетики человека
1.3.2.35. Генные (молекулярные)
болезни
История исследований болезней обмена
веществ у человека убедительно показывает, что пионерами в данной области были
клиницисты, что описали в конце прошлого и начале нынешнего столетия
некоторые из этих патологий. Знаменитая концепция А. Гаррода о врожденные нарушения
обмена как о определенные метаболические блоки, сформулированная еще в 1908 г., стимулировала
биохимические исследования в этом направлении, особенно широкое развитие они
получили в последние 2-3 десятилетия. До нынешнего времени накоплен огромный
материал с биохимической характеристики многих болезней обмена, без чего
невозможна четкая диагностика и лечение. Однако, как и ранее, больные с дефектами обмена на первых этапах обследования
попадают в первую очередь в поле зрения клиницистов различных профилей, перед которыми
встает очень трудная задача - отнести ту или иную патологию к определенной болезни
обмена и организовать комплексное клинико-биохимическое обследование.
Многие заболевания обусловлены
мутациями, которые изменяют генетическую конституцию человека, что приводит к
нарушение нормального функционирования организма. Уже выявлено около 600
наследственных нарушений метаболизма, но только для 105 из них установлен точный
уровень "метаболического блока" и характер дефекта (рис. 1.144).
Исследователи продолжают идентифицировать все новые и новые заболевания из этой
группы.
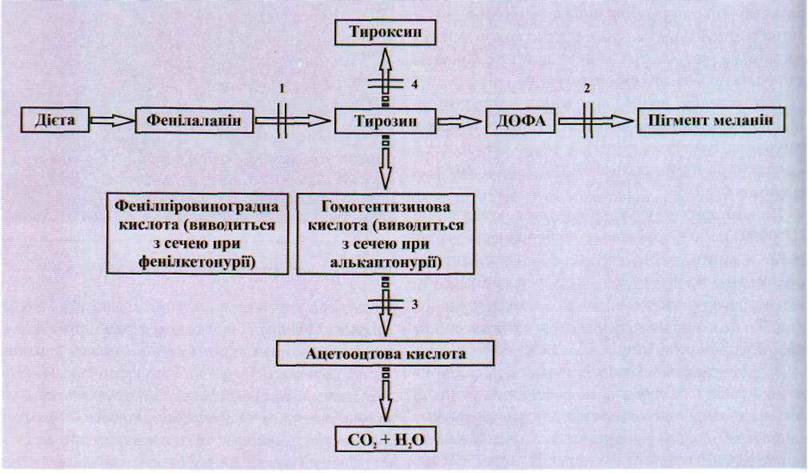
Рис. 1.144. Биохимические
"блоки" при наследственных нарушениях обмена аминокислот:
1 - фенилкетонурия; 2 - альбинизм; 3
- алькаптонурія; 4 - врожденная недостаточность тироксина (дисгормоногенез).
На основании данных современной
биохимической генетики можно объяснить, каким образом генетическая информация транслируется
при синтезе белков со специфическими метаболическими или структурными
особенностями. Все названи мутации могут приводить как к
нарушение первичной структуры белка, так и к изменению количества синтезированного
специфического белка. Если процесс нарушен врожденным дефектом метаболизма,
имеет существенное значение для здоровья и если степень изменений достаточный для проявления
патологического процесса, то могут проявляться клинические признаки. Некоторые генетические
изменения не сопровождаются клиническими проявлениями и лишь определяют полиморфизм, что
отличает одного индивида от другого. Другие изменения могут проявляться лишь при
определенных условий, которые на протяжении всей жизни могут и не возникнуть. Наконец, вероятные
такие генетические нарушения, которые вызывают заболевания, выраженность которого
колеблется от очень умеренных проявлений до состояний, приводящих к летальному исходу. В большинстве случаев
врожденные нарушения обмена веществ с клиническими последствиями проявляются (или могут
быть обнаруженными) в период новонароджуваності. Такие младенцы сразу после
рождения конечно выглядят здоровыми, однако признаки патологии, такие как
летаргия, затруднение три кормления, судороги, рвота и др., могут проявиться в
них уже через несколько часов. Некоторые нарушения метаболизма могут остаться
нераспознанными в период новонароджуваності и диагноз может быть поставлен
только через несколько месяцев и даже лет. Ранние клинические проявления обычно
неспецифические и могут быть отнесены к перинатальной патологии. Врожденное
нарушение обмена веществ должно рассматриваться как возможный состояние у любого ребенка с
одним из указанных клинических проявлений:
• неопределенное отставание
умственного, двигательного развития, судороги;
• необычный запах, в частности, под
время острого заболевания;
• інтермітуючі эпизоды
необоснованной рвоты, ацидоза, нарушений психики, кома;
• почечная колика, гепатомегалия.
Классификация молекулярных нарушений
обмена веществ
1. Нарушения метаболизма
аминокислот:
1.1. Фенилаланина (фенилкетонурия);
1.2. Тирозина (тирозинемия,
алькаптонурія);
1.3. Метионина (гомоцистинурия);
1.4. Цистина (цистинурия);
1.5. Триптофана (болезнь Хартнупа,
триптофанемія и др.);
1.6. Лейцина (болезнь кленового
сиропа);
1.7. Гистидина
(гістидинурія, гистидинемия) и других аминокислот.
2. Нарушения метаболизма
углеводов:
2.1. Галактозы (галактоземия);
2.2. Фруктозы (фруктоземия);
2.3. Гликогена (гликогенозы);
2.4. Дисахаридозні
энтеропатии (синдром мальабсорбции углеводов).
3. Наследственные болезни
обмена соединительной ткани:
3.1. Мукополисахаридозы;
3.2. Болезнь Марфана.
4. Наследственные болезни обмена липидов:
4.1. Гиперлипопротеинемии;
4.2. Сфінголіпідози (болезнь Німанна
- Пика);
4.3. Гангліозидози (болезнь Тея -
Сакса).
5. Наследственные болезни
порфиринового обмена (порфирии).
6. Энзимопатии
желчно-пигментного обмена (болезнь Жильбера).
7. Энзимопатии
панкрео-инсулярного гормоно-синтеза:
7.1. Муковисцидоз;
7.2. Урожденная
отсутствие энзимов поджелудочной железы;
7.3. Болезнь Вильсона - Коновалова;
7.4. Целиакия.
8. Энзимопатии биосинтеза гормонов.